Ionen - Kapillar - Elektrophorese
"Leben auf schleimigem Fuß" Ökophysiologie limnischer Gastropoden |
|---|
Ionen - Kapillar - Elektrophorese |
|
Identifizierung der Ionen:
Wie bereits erwähnt, passieren die Ionen nach unterschiedlichen Zeiträumen den Detektor.
Von den Standardlösungen und ihren Verdünnungen kann man nun die
relativen Laufzeiten bestimmen: Man setzt dabei die Laufzeiten der unterschiedlichen
Ionen ins Verhältnis zu der niedrigsten vorkommenden Laufzeit. Bei den Anionen hat
Chlorid die niedrigste Laufzeit bei den Kationen Ammonium. Dazu muß man zunächst dieses Ion in einer Probe
identifizieren, erst dann können die relativen Laufzeiten bestimmt und mit denen der Standards verglichen werden.
Ist das Ion in einer Probe nicht vorhanden, kann man es in einem zweiten Durchlauf hinzufügen und ebenso verfahren wie vorher.
Man kann nicht die absoluten Laufzeiten verwenden, da sich diese mit zunehmender Versuchsdauer ändern. Durch die steigende Temperatur, die von der angelegten Spannung herrührt, sinkt die Viskosität der Lösung und die Mobilität der Ionen steigt.
Diese Methode ist relativ zuverlässig. Kann man einen Peak dennoch nicht genau identifizieren, kann man die qualitative Additionsmethode anwenden:
Hierbei fügt man den für den Peak vermuteten Stoff der Probe zu und führt den
Versuch noch einmal durch. Ist der Peak größer als beim ersten Durchlauf, lag man mit
seiner Vermutung richtig. Befinden sich an der Stelle des unbekannten Peaks nun zwei Peaks, sind
die Stoffe nicht identisch und das Verfahren muß wiederholt werden.
Man kann die Methoden auch kombinieren:
Liegt die relative Laufzeit eines Peaks nur ungefähr bei einer der Standardlösung, kann man zur Kontrolle die qualitative Additionsmethode anwenden.
Die von uns identifizierbaren Ionen sind:
Anionen: Kationen:
Chlorid Natrium
Sulfat Kalium
Nitrat Calcium
Fumarat Magnesium
Malat Ammonium
Succinat
Acetat
Lactat
Propionat
Phosphat
Carbonat
Citrat
Quantifizierung der Ionen:
Die Fläche unter den Peaks der Ionen ist proportional zur in der Probe enthaltenen Stoffmenge.
Mit Hilfe der Standardlösungen ermittelt man Regressionsgeraden der Flächen über der
Konzentration mit der dazugehörigen Formel für jedes Ion. Nach Umstellen der Formel kann
man aus den erhaltenen Flächen die Konzentration des Ions in der Probe berechnen.
Es kommt vor, daß durch den Puffer in der Probe enthaltene Ionen "weggefangen" werden und
die erhaltene Konzentration nicht mit der tatsächlichen in der Probe übereinstimmt. Man
kann zur Ermittlung der tatsächlichen Konzentration nacheinander bekannte, unterschiedliche
Konzentrationen des Ions hinzugeben und die Fläche unter den Peaks ermitteln. Trägt man
die Fläche über den zugegebenen Mengen des Ions auf und zieht eine Gerade, so ist ihr
Schnittpunkt mit der x-Achse die tatsächliche in der Probe enthaltene Konzentration mit
negativem Vorzeichen.
Atom-Absorptions-Spektrometrie |
|
Die Atomambsorptionspektrometrie (AAS) dient der quantitativen Analyse eines Elementes in gelöster Form. Das Prinzip der AAS gehorcht einem von Kirchhoff formulierten Gesetz, welches besagt, daß jeder Stoff Strahlung der Wellenlänge, die er selbst emittiert, auch absorbieren kann. Bestrahlt man also Atome einer zu quantifizierenden Substanz mit Licht aus einer Strahlungsquelle des interessierenden Elementes, so können die Atome der Lösung etwas von den einstrahlendem Licht absorbieren. Der Extinktionsunterschied zwischen dem Licht vor und nach passieren der Lösung kann photometrisch gemessen werden und steht in Relation mit der Atomkonzentration in der Lösung. Das Element in der Lösung muß in Form von freien Atomen vorliegen, hierfür wird die Lösung vorab zerstäubt und in einer Flamme in ihre atomaren Teilchen zerlegt. Das Einstrahlungslicht trifft in der Flamme auf die freien Atome.
Apparatur:
Zum Aufbau der AAS gehören also
Als Strahlungsquelle benutzten wir ein Hohlkathodenlampe. Die Kathode besteht aus dem anzuregendem Element. Sie ist von einem gasgefüllten Glaszylinder umgeben. Bei Anlegen einer Spannung (bis 600 V) zwischen den Elektroden bilden sich in dem Glaszylinder an der Kathode positive Gasionen, welche auf die Kathodenoberfläche schlagen und somit Atome des Elementes herauslösen und zur Strahlung anregen.
Zur Atomisierung der Probe benutzten wir ein Gemisch aus Acetylen als Brenngas und Luft als Oxidant. Um die Anzahl der absorbierenden Atome zu maximieren, empfiehlt sich die Benutzung eines Schlitzbrenners, welcher durch eine Flammenbreite bis zu 10 cm einen langen Weg der Strahlung durch die Flamme ermöglicht. Eine Spiegeleinrichtung, welche den Strahlengang mehrmals durch die Flamme passieren läßt, erhöht ebenfalls die Wahrscheinlichkeit, daß ein freies Atom ein Photon absorbiert. Die Temperatur der Flamme sollte nicht zu hoch sein, um die mögliche Eigenemission durch freie, zu stark angeregt Atome zu verhindern; sie würde der zu messenden Absorption entgegenwirken und somit zu niedrige Extinktionswerte bewirken. Bedingt ist die Flammentemperatur durch die Wahl der Brenngase.
Als Filter wird ein Spektralphotometer benutzt. Einfallende kontinuierliche Strahlungen werden in diskrete Spektren zerlegt. Durch Regulierung der Spaltbreite vor dem Eingang des nachgeschalteten Detektors entscheidet man sich für die Spektren des zu analysierenden Elementes und schließt somit eventuelle Störungen oder Verunreinigungen in der Apparatur aus.
In dem Detektor erfolgt die Verstärkung der gemessenen Energiedifferenz und die Umwandlung in ein Anzeigesignal.Wir beschränkten uns in der ersten Woche auf die Nutzung der AAS hauptsächlich zur Erstellung einer Eichkurve für spätere Calziumbestimmungen.
Einstellungen am Gerät: (Skript Seite 30)
Verbrennungskalorimetrie |
|
Die Temperaturerhöhung des Gefäßes wird von einem Schreiber aufgezeichnet. Man muß nicht warten bis die entstehende Kurve vollständig aufgezeichnet wird, sondern man kann nach dem Erreichen des Maximums den Versuch unterbrechen und den Energiegehalt rechnerisch ermitteln.Vor dem Zünden muß man die Bombe in dem Kalorimeter einige Minuten ruhen lassen und auf dem Schreiber sollte eine mehr oder weniger gerade Linie von 2-3 cm zu beobachten sein.
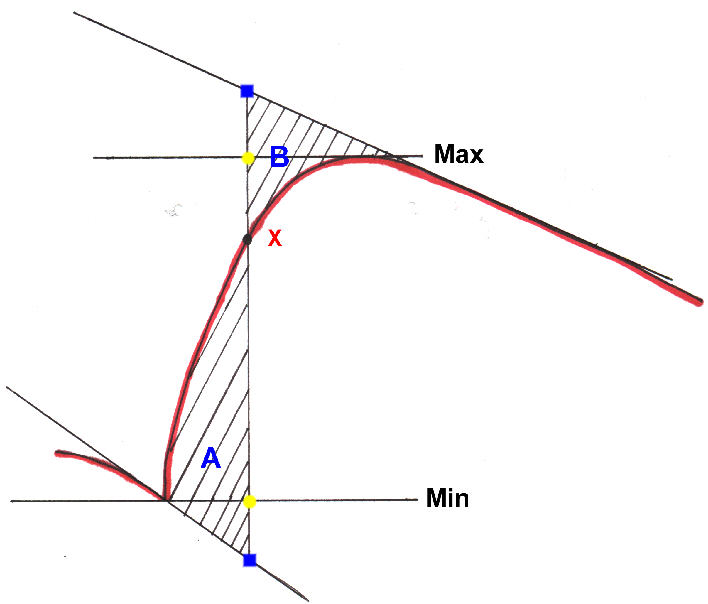
Zur Auswertung legt man eine Gerade durch die Anfangssteigung (vor dem Zünden) und
eine durch die Endsteigung der aufgezeichneten Kurve. Durch einen bestimmten Punkt X auf
der Kurve fällt man ein Lot. Die Länge (in mV) dieses Lots zwischen seinen
Schnittpunkten mit der Anfangs- bzw. Endsteigung entspricht dem Energiegehalt
der Probe. Um den Zusammenhang zwischen den aufgezeichneten Spannung und der
tatsächlichen Energie zu erhalten, erstellt man eine Kalibrierungsgerade mit Hilfe von
Benzoesäure, die eine Verbrennungswärme von 26 470 J/g besitzt (Kalibrierung).
Das
gefällte Lot muß so liegen, dass die Flächen
zwischen der Kurve und der Anfangssteigung (A) gleich der derjenigen zwischen
Kurve und Endsteigung (B) ist. Zur Vereinfachung wird dieser Flächeninhalt nur
bei den Benzoesäuren direkt gemessen. Man misst dann auf welcher Höhe er auf
der Min-Max-Gerade liegt und gibt diese Größe in % des Maximums an.
Aus dem Mittelwert kann man für jede weitere Messung schnell den Punkt X auf der Kurve bestimmen.
Sowohl der Kalibrierfaktor als auch die Lage des Punktes X sind
abhängig von der verwendeten Bombe und muss für jede extra ermittelt werden.
Differenz-Thermo-Analyse |
|
Im Anhang befindet sich eine Tabelle mit näheren Erläuterungen zu den verwendeten Chemikalien